Production #450
Стартовые системы
| Status: | In Progress | Start date: | 14.09.2021 | ||
|---|---|---|---|---|---|
| Priority: | Normal | Due date: | 30.10.2021 | ||
| Assignee: | % Done: | 50% | |||
| Category: | - | ||||
| Target version: | - |
Description
Подготовить стартовую систему комплекса PGM c двумя вариантами субстратов. Под стартовой системой подразумевается залитая водой GRO (вода вычищена) с топологией.
Как делать параметры для малых молекул (альтернативы):
1) https://www.bio2byte.be/acpype/ - выбрать GAFF2
2) https://github.com/openforcefield/openff-toolkit - выбрать Parsley или Sage, если есть. Разобраться, это не однокнопочное решение!
3) https://github.com/openforcefield/bespoke-fit
Нам как лабе будет очень полезно, если ты потратишь время и освоишь подготовку параметров для малых молекул вариантами 2 и 3. С 2 тебе может помочь Юлия Беляева, она уже так делала. 3 для нас совсем новый.
Тебе может понадобиться поставить свою миниконду под своего пользователя на срв и научиться делать энвайронменты.
фейл.jpg (100 KB)
{kind=link}
3ezn_rdpg.gro (329 KB)
RDPG_CHARMM.prm (2.39 KB)
RDPG_GMX.itp (14.1 KB)
Молекулярное моделирование каталитического механизма фосфоглицерат мутазы.png (69 KB)
{kind=link}
3GP5_ph_bond_1.pdb (165 KB)
History
#1
 Updated by Anastasia Rachkova about 4 years ago
Updated by Anastasia Rachkova about 4 years ago
- Due date changed from 27.09.2021 to 02.10.2021
Сравнить по литературным данным тулы для генерации параметров для малых молекул и выбрать тот, что больше подходит.
#2
Updated by Anastasia Rachkova about 4 years ago
- Due date changed from 02.10.2021 to 04.10.2021
- % Done changed from 0 to 50
Решено пока оставить Bespoke-fit в покое и сделать все с помощью GAFF.
#3
Updated by Anastasia Rachkova about 4 years ago
Сделаны параметры для субстратов - обоих стереоизомеров. Далее нужно
1) сделать системы комплексов
2) сделать минимизацию
#4
Updated by Anastasia Rachkova about 4 years ago
За основу фермента взяла белок из 3EZN буркходелии, очистила его от воды и гликолей.
Хотела бахнуть pdb2gmx (поле amber03 и модель PIP3P - как делали в статье, в которой моделировали С-конец) , но вылезла ошибка:
Fatal error:
Residue 38 named GLN of a molecule in the input file was mapped
to an entry in the topology database, but the atom CG used in
that entry is not found in the input file. Perhaps your atom
and/or residue naming needs to be fixed.
Решение ее оставляю на день, возможно, имеет смысл просто взять другой pdb.
#5
 Updated by Alexander Zlobin about 4 years ago
Updated by Alexander Zlobin about 4 years ago
- Status changed from New to In Progress
Такс
Во-первых, поле 03 очень древнее - на это намекает цифра 03, то бишь 2003 год. Что брать - ff19sb. Как это делать:
export GMXLIB=/home/domain/data/zlobin/gmxlib
И потом pdb2gmx -ff amber19sb
Во-вторых, по поводу этой самой ошибки
Посмотрите внимательно на остаток, идущий 38-ым в PDB. Вероятно, каких-то атомов попросту не хватает (его номер исходно может быть и не таким, но Громаксу на это пофигу, ему важен именно номер по порядку. Чтобы не запутаться, можно исходно перенумеровать стартовый PDB, чтобы номера остатков шли с 1. gmx editconf -f <input.pdb> -o <output.pdb> -resnr 1. Как с этим бороться - либо руками в PyMol (Wizard > Mutagenesis и бахнуть ту аминку, которая там и должна быть. Ротамер выбрать на свой вкус). Либо есть автоматические инструменты, типо PDBFixer для питона.
В-третьих, кристаллическую воду не надо убирать. Потом ждать, что сгенерированная зальется в места, где должна быть структурная вода - вечность.
#6
Updated by Anastasia Rachkova about 4 years ago
Взяла новое поле и пофиксила глутамин 38 (pdbfixer'ом), команда:
gmx pdb2gmx -ff amber19sb -f 3ezn.pdb -o 3ezn_processed.gro
Вылезла ошибка "Fatal error:
Atom HB3 in residue MET 1 was not found in rtp entry NMET with 19 atoms
while sorting atoms." Буду игнорировать водороды:
gmx pdb2gmx -ff amber19sb -f 3ezn.pdb -o 3ezn_processed.gro -ignh - сработало!
Надо проверить, что там вообще наделалось: в нормальной ли (неанионной форме аспартат и глутамат -> в pdb2pqr посчитать pKa остатков) и в какой таутомерной форме гистидины, хотя бы внутренние.
#7
Updated by Alexander Zlobin about 4 years ago
Привет, апдейтни по подготовке систем, пожалуйста. Когда смогу их посмотреть и обсудить?
И следи за своевременным обновлением дедлайнов с пояснениями.
#8
Updated by Anastasia Rachkova about 4 years ago
- Due date changed from 04.10.2021 to 30.10.2021
У меня небыстро продвигается с ручной обработкой гистидинов и аспарагинов с глутаминами. Хочу к 30 ноября доделать наконец и попробовать запустить моделирование, посмотреть, не сломается ли ничего.
Если можно, обсудила бы с вами, что получилось, в понедельник или вторник (1 или 2 ноября).
#9
Updated by Alexander Zlobin about 4 years ago
Подготовка корректных систем - половина успешности моделирования, так что да, каким бы ни было унылым, делать приходится.
Да, конечно, как только дойдете до добавления ионов, дайте ссылку на эти файлы.
#10
Updated by Anastasia Rachkova about 4 years ago
У меня какой-то фейл с тем, чтобы вращать остатки амидов, делаю это не по-русски. Вы не подскажете еще раз, пожалуйста, что вы использовали, чтобы только их поворачивать?
#11
Updated by Alexander Zlobin about 4 years ago
Режим editing в Pymol
Ctrl + ПКМ, щелчок по связи и зажать - будет вращение той группы, к которой ближе вы щелкнули.
#12
Updated by Anastasia Rachkova about 4 years ago
Спасибо! pdb сделала, двигаюсь дальше.
#13
Updated by Anastasia Rachkova about 4 years ago
- File фейл.jpg added
- File 3ezn_rdpg.gro added
- File RDPG_CHARMM.prm added
- File RDPG_GMX.itp added
Сделала общий файл "3ezn_rdpg.gro" с координатами.
Потом делала ячейку и заливала ее водой:
gmx editconf -f 3ezn_rdpg.gro -o 3ezn_rdpg__cell.gro -bt cubic -d 1.0
gmx solvate -cp 3ezn_rdpg__cell.gro -cs spc216.gro -p topol.top -o 3ezn_rdpg_solv.gro
Не уверена, что стоило делать ее такой формы, в мануале был додекаэдр, но мне показалось, что это может быть незачем, в статьях (https://www.frontiersin.org/articles/10.3389/fmolb.2018.00065/full
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6438627/) делали кубик и я попробовала тоже.
gmx solvate -cp 3ezn_rdpg__cell.gro -cs spc216.gro -p topol.top -o 3ezn_rdpg_solv.gro
Дальше хотела бахнуть ионы:
gmx grompp -f ions.mdp -c 3ezn_rdpg_solv.gro -p topol.top -o ions.tpr
Но потерпела поражение.
Может, использовала какие-то совсем не те файлы...
- Оставлю себе тут пару ссылок на статьи про моделирование ***
[[https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4030948/]]
[[https://tbiomed.biomedcentral.com/articles/10.1186/1742-4682-11-52]]
#14
Updated by Alexander Zlobin about 4 years ago
А путь-то какой к файлам? Где они лежат?
#15
Updated by Anastasia Rachkova about 4 years ago
PDB Burchodelii 3ezn - удалила одну из цепей, гликоли удалила, воду оставила.
pdbfixer 3ezn_Achain.pdb --output=3ezn.pdb --add-atoms=all
gmx pdb2gmx -ff amber19sb -f 3ezn.pdb -o 3ezn_processed.gro -ignh
gmx editconf -f 3ezn_processed.gro -o 3ezn_processed.pdb
Дальше опять ручками поправляла гистидины, аспарагины и глутамины. Даже полезно второй раз с этим потренироваться.
alter (sele), resn="HID"
gmx editconf -f 3ezn_his.pdb -o 3ezn_his.gro
В файле 3ezn_rdpg.gro совместила белок и лиганд, изменила количество строк. Также внесла изменения в topol.top (только itp файл, никаких prm).
gmx editconf -f 3ezn_rdpg.gro -o 3ezn_rdpg_cell.gro -bt cubic -d 1.0
gmx solvate -cp 3ezn_rdpg_cell.gro -cs spc216.gro -p topol.top -o 3ezn_rdpg_solv.gro
Тут повозилась с тем, что у меня itp файл был немножко кривоват - исправила вручную.
gmx grompp -f ions.mdp -c 3ezn_rdpg_solv.gro -p topol.top -o ions.tpr - ошибка,
"number of coordinates in coordinate file (3ezn_rdpg_solv.gro, 121106)
does not match topology (topol.top, 4276)"
#16
Updated by Anastasia Rachkova about 4 years ago
Итоги встречи 10 ноября:
-текстовые файлы надо всегда смотреть и проверять ручками
-чтобы юзать поле, можно просто один раз за сессию сделать export нужной папки
-для новой попытки создать новую папку без ненужных файлов
Про планы на ближайшее время:
-взять pdb 3GP5, в котором уже есть 2,3-дифосфоглицерат. На первый взгляд, его заряд -5 и водородов нигде нет. Туда подобавлять воду. Потом в текстовом файле поменять гистидин на H2E.
-мне получить с помощью acpype параметры для 2-PG, 2,3-PG, 3-PG. Можно рисовать в Avogadro, есть на сервере.
-А.С. взять параметры для фосфогистидинов и переделать их под поле 19 года.
#17
Updated by Alexander Zlobin about 4 years ago
Фосфорилированные остатки в поле добавил. Теперь достаточно остатку иметь нужное название и атомы фосфора и фосфорильных кислородов. Варианты гистидинов: H1E, H2E, H1D, H2D. Буква - к какому азоту крепится фосфат, цифра - заряд на фосфате. Нам нужен H2D.
#18
Updated by Anastasia Rachkova about 4 years ago
Для 2,3-фосфоглицератов заряд ставила -5, для остальных -4. Рисовала все ручками в Авогадро.
Файлы лежат в папке /home/domain/rachkovanastya/PGM_start
#19
Updated by Anastasia Rachkova about 4 years ago
Файл с фосфогистидином сделала, но на команде pdb2gmx -ff amber19sb -f 3ezn.pdb -o 3ezn_processed.gro -ignh ложится, пдб фиксером не фиксится. попробую что-то сделать еще.
лежит в PGM_start, 3PG5_ph_his.pdb
#20
Updated by Alexander Zlobin about 4 years ago
С чем конкретно ложится?
#21
Updated by Anastasia Rachkova almost 4 years ago
Забавно, когда делала первый раз - у него была ошибка на Met1, сейчас такая:
Fatal error:
Atom O3 in residue HIS 9 was not found in rtp entry CHIE with 18 atoms
while sorting atoms.
#22
Updated by Alexander Zlobin almost 4 years ago
Ну он тут все говорит прямо. В его рецепте для остатка HIE (в данном случае Ц-концевого HIE - кстати, проверь, должен ли он быть Ц-концевым, может там закрался лишний TER в файл) нет атома с именем O3. И действительно не должно быть.
При этом в PDB файле он, видимо, есть.
Открываю и вижу:
ATOM 72 N HIS A 9 4.651 -27.787 32.625 1.00 13.38 N
ATOM 73 CA HIS A 9 4.325 -27.748 34.022 1.00 14.08 C
ATOM 74 C HIS A 9 2.943 -28.347 34.257 1.00 14.64 C
ATOM 75 O HIS A 9 2.086 -28.343 33.390 1.00 13.13 O
ATOM 76 CB HIS A 9 4.350 -26.304 34.565 1.00 14.62 C
ATOM 77 CG HIS A 9 3.346 -25.393 33.921 1.00 14.21 C
ATOM 78 CD2 HIS A 9 3.446 -24.561 32.849 1.00 14.41 C
ATOM 79 ND1 HIS A 9 2.054 -25.288 34.370 1.00 16.06 N
ATOM 80 CE1 HIS A 9 1.403 -24.419 33.618 1.00 17.01 C
ATOM 81 NE2 HIS A 9 2.217 -23.975 32.675 1.00 13.92 N
TER
HETATM 82 O3 HIS A 9 3.248 -21.563 31.620 1.00 16.33 O
HETATM 83 O2 HIS A 9 2.001 -24.090 30.109 1.00 17.09 O
HETATM 84 O1 HIS A 9 0.261 -22.293 32.290 1.00 17.35 O
HETATM 85 P HIS A 9 1.669 -22.473 31.040 1.00 17.72 V
Видимо, это должен быть быть фосфогистидин. Но он называется не HIS и не HIE. Нужно дать ему его имя (H2E, H2D, что-то такое же было?)
Во-сторых, между атомами закрался TER, что командует громаксу разорвать цепь. Его нужно отсюда убрать.
#23
Updated by Anastasia Rachkova almost 4 years ago
Интересно, когда я пофиксила pdbfixer'ом созданный мною файл, там реально получился какой-то треш. Сейчас пробую сделать заново, но пока там тирозином и не пахнет, откуда он вообще взялся.
#24
Updated by Alexander Zlobin almost 4 years ago
Каким тирозином?... Я что-то совершенно запутался. Не надо использовать никакой pdbfixer, он ничего не знает про фосфогистидины.
#25
Updated by Anastasia Rachkova almost 4 years ago
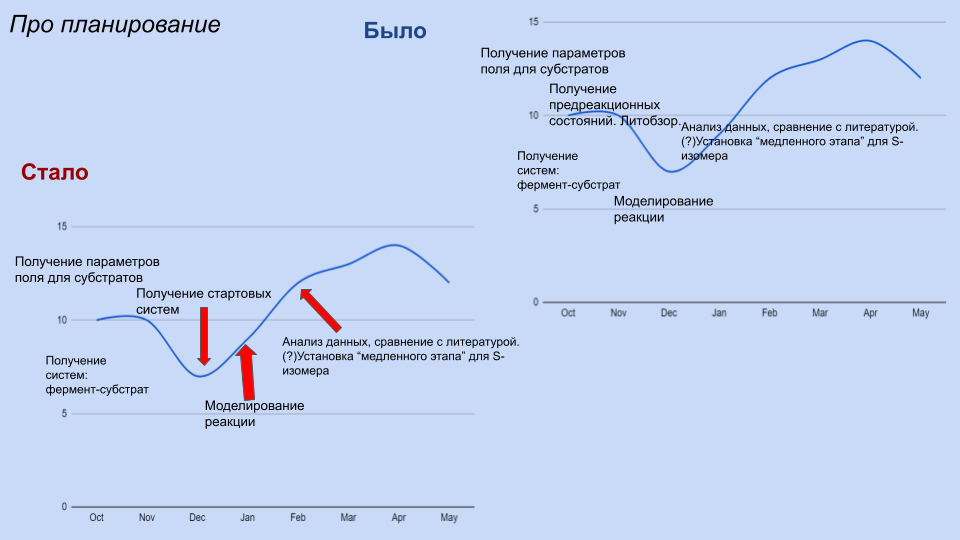
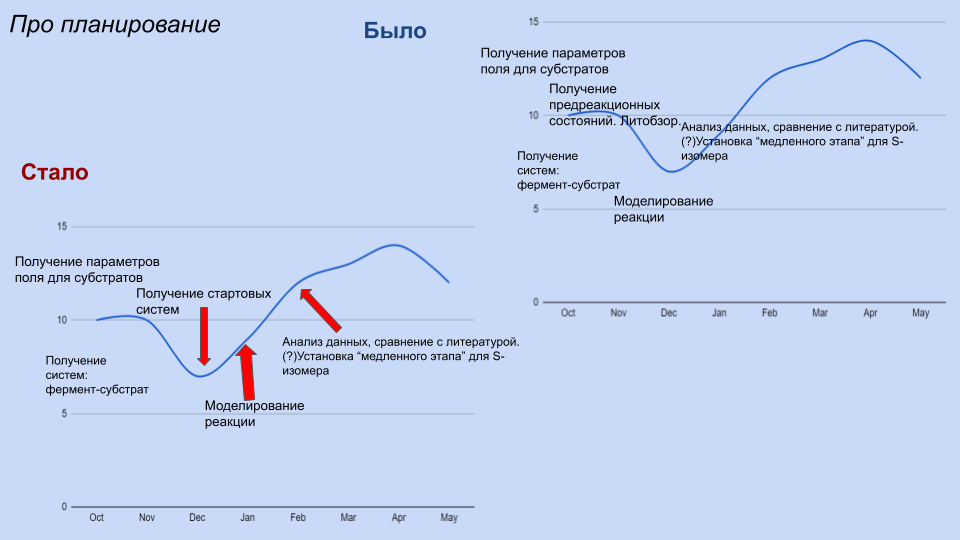
Добрый вечер! Пишу про сроки проекта. На самом деле я не так уж и отклонилась от исходного плана: в декабре сделать стартовые системы для обоих изомеров. В январе провести собственно моделирование реакции (двух вариантов), переделать, если все взорвется и сломается. В феврале - анализ результатов, сравнение скорости для двух изомеров. Я думаю, что успеть это сделать реально, но необходимо не допускать больше сидения на одном этапе месяц без продвижений.
Такое замедление в ноябре и начале декабря можно объяснить тем, что я уезжала в Сириус, а потом много тупила и еще и не спрашивала сразу, если нужна была помощь.
Меры пресечения такого:
Два раза в неделю в независимости от наличия вопросов писать тут (или в другой системе, которая будет) отчет по выполненной части работы (как минимум для себя).
Задавать вопросы вам или в общую беседу сразу по мере поступления, если не могу их решить сама за вечер.
#26
Updated by Anastasia Rachkova almost 4 years ago
- File 3GP5_ph_bond_1.pdb added
А еще я разобралась-таки с исходным pdb. Исправила название атома, убрала этот TER и сделала фосфатную группу частью основания. Но gromax снова выдал ошибку. Кажется, у него проблемы с тем, что он не может в своих файлах найти основание H2E.
Fatal error:
The residues in the chain MET1--ALA248 do not have a consistent type. The
first residue has type 'Protein', while residue H2E9 is of type 'Other'.
Either there is a mistake in your chain, or it includes nonstandard residue
names that have not yet been added to the residuetypes.dat file in the GROMACS
library directory. If there are other molecules such as ligands, they should
not have the same chain ID as the adjacent protein chain since it's a separate
molecule.
#27
Updated by Anastasia Rachkova almost 4 years ago
С тирозином это я и сама запуталась, извините. А вот с O3 не поняла, там же действительно 3 кислорода, почему не должно быть O3?
#28
Updated by Alexander Zlobin almost 4 years ago
Потому что у остатка HIE нет никаких кислородов. Они есть у остатка H2E. Это совсем разные остатки.
Сейчас ошибка говорит, что H2E не является белком. Как он это понимает? Есть в папке на один уровень выше папки с полем файлик residuetypes.dat. Там нужно прописать то. что является белком. Я это действительно не прописал раньше. Сейчас добавил, попробуй заново.
По поводу плана -- получим модели до 30 декабря, никаких проблем. Но да, нужно не терять темпа работы. Сириус меня вообще никак не касается, это вопрос лично твоих приоритетов. В общий чатик, конечно, стоит писать) А еще читать, что там есть, очень много карьерно значимых вещей там обсуждается. Все, что за гранью конкретной курсовой, но про карьеру.
Я также прошу выделить на работу в лаборатории конкретные временные слоты и сообщить их мне. Например "понедельник, 18-20, среда, 15-16", в таком духе. Один набор до конца января, и второй -- на следующий семестр, как станет известно расписание.
#29
Updated by Anastasia Rachkova almost 4 years ago
Точное время для лабораторной работы до конца января:
Понедельник 12:30 - 14:00
Среда 14:00 - 16:30
Суббота либо 14:00 - 16:00, либо 19:00 - 21:00
#30
Updated by Anastasia Rachkova almost 4 years ago
Попробовала еще раз, ошибка следующая:
Fatal error:
Atom O1 in residue H2E 9 was not found in rtp entry H2E with 21 atoms
while sorting atoms.
Видимо, у вас (то бишь в файлах с параметрами поля) атомы кислорода в фосфатной группе называются как-то по-другому?
#31
Updated by Alexander Zlobin almost 4 years ago
Вы можете сами такие вещи смотреть.
Отсылаю к этой части обсуджения: export GMXLIB=/home/domain/data/zlobin/gmxlib
Это путь к полям. Внутри есть папка amber19sb, в ней есть aminoacids.rtp
внутри него есть такие строчки:
[ H2E ]
[ atoms ]
N N -0.5163 1
H H 0.2936 2
CA CX 0.330527 3
HA H1 0.031846 4
CB CT -0.410313 5
HB1 HC 0.101457 6
HB2 HC 0.101457 7
CG CC 0.240309 8
ND1 NA -0.372805 9
HD1 H 0.355424 10
CE1 CR 0.039056 11
HE1 H5 0.212475 12
NE2 NA -0.052173 13
CD2 CW -0.225382 14
HD2 H4 0.2656 15
P P 1.349034 16
O1P OO -0.899504 17
O2P OO -0.899504 18
O3P OO -0.899504 19
C C 0.5366 20
O O -0.5819 21
Соответственно атомы кислорода это O1P O2P O3P
#32
Updated by Anastasia Rachkova almost 4 years ago
О, спасибо большое за инструкцию!
Я сделала рабочий файл-таки, пришлось сначала фиксить его без фосфогистидина, а потом уже добавлять фосфогистидин.
#33
Updated by Anastasia Rachkova almost 4 years ago
Локально (только на этой неделе) переношу время работы над проектом с сегодня (среда) но четверг.
#34
Updated by Anastasia Rachkova almost 4 years ago
Добрый вечер!
Я продвинулась дальше, но снова наткнулась на неочевидную ошибку при запуске команды:
gmx grompp -f ions.mdp -c 3GP5_solv.gro -p topol.top -o ions.tpr
_ERROR 1 [file ffbonded.itp, line 2373]:
Unknown bond_atomtype nN
There was 1 note
-------------------------------------------------------
Program: gmx grompp, version 2018.1
Source file: src/gromacs/gmxpreprocess/toppush.cpp (line 943)
Fatal error:
There was 1 error in input file(s)_
Эта строка в файле выглядит вот так:
[ dihedraltypes ] ; improper
.....
; lipid17
cA cA cB cB 4 180.0 4.6024 2
cA cB cB hB 4 180.0 4.6024 2
cA oO cC oO 4 180.0 4.6024 2
cB cD cB hB 4 180.0 4.6024 2
cD oC cC oS 4 180.0 43.932 2
+ cA cC nN hN 4 180.0 4.6024 2+
cD nN cC oC 4 180.0 4.6024 2
#35
Updated by Anastasia Rachkova almost 4 years ago
Этот момент в файле вроде не касается даже аминоксилот и прочего, но я как-то не рискнула просто удалить эту строчку в своей копии файла, кажется, это не очень хорошо.
#36
Updated by Anastasia Rachkova almost 4 years ago
Важные моменты:
В файле complex.gro лучше сразу ставить атомы лиганда сразу после белка и перед водой, все атомы должны идти друг за другом, а атомы лиганда еще и в том же порядке, что в файле лигагд.itp. Еще в 1ой строке не забываем менять кол-во атомов всего (плюсуем атомы лиганда)
В файл topol.top добавляем
; Include ligand topology
#include "RthreePG_GMX.itp"
в начале, сразу после
; Include forcefield parameters
#include "amber19sb.ff/forcefield.itp"
А еще в молекулс добавляем лиганд
; Compound #mols
Protein_chain_A 1
R3PG 1
SOL 27
При исполнении gmx grompp -f ions.mdp -c 3GP5_cell.gro -p topol.top -o ions.tpr возникла проблема с тем, что атом лиганда h1 уже был записан в поле ранее, я просто переименовала его в файлах RthreePG_GMX.itp и complex.gro
gmx genion -s ions.tpr -o 3GP5_ions.gro -p topol.top -pname NA -nname CL -neutral - выбрала группу 15 (SOL)
#37
Updated by Anastasia Rachkova almost 4 years ago
При выполнении
gmx grompp -f em.mdp -c 3GP5_ions.gro -p topol.top -o em.tpr
возникла сдедующая ошибка:
Fatal error:
Something is wrong in the coordinate formatting of file 3GP5_ions.gro. Note
that gro is fixed format (see the manual)
Наверное, единственное, где могли бы быть проблемы, это последние строки, которые обновлялись при добавлении ионов:
267SOL OW 3992 5.418 3.642 2.514
267SOL HW1 3993 5.500 3.642 2.572
267SOL HW2 3994 5.337 3.642 2.572
268SOL OW 3995 5.746 4.095 2.983
268SOL HW1 3996 5.828 4.095 3.041
268SOL HW2 3997 5.746 4.176 2.925
269SOL OW 3998 6.368 4.124 4.160
269SOL HW1 3999 6.450 4.124 4.217
269SOL HW2 4000 6.368 4.042 4.102
270NA NA 4001 5.503 2.427 3.140
271NA NA 4002 4.199 3.078 3.688
272NA NA 4003 5.995 3.288 3.706
273NA NA 4004 5.808 2.242 4.209
274NA NA 4005 5.324 4.035 4.129
275NA NA 4006 5.000 1.807 3.258
276NA NA 4007 4.777 3.340 3.912
8.88820 8.88820 8.88820
Путь к этому файлу в моей папке: /home/domain/rachkovanastya/PGM_start/3GP5_ions.gro
#38
Updated by Anastasia Rachkova almost 4 years ago
Что есть:
прочитала статьи.
для R-изомера еще раз заново сделала файл белка, поправила руками Asn, Gln, His. Сделала комплекс, добавив лиганд с координатами из его комплекса с белком. Сделала ячейку (d = 0,5), залила водой, добавила ионы. Минимизация энергии пошла, но прервалась из-за неправильного положения какой-то молекулы воды (я предполагаю, какой, попробую днем поправить).
для S-изомера получила файл белка, начала делать комплекс, обнаружила, что неправильно посчитала параметры для лиганда - надо пересчитать и доделать.
Соответственно, днем 10ого - поправить воду в комплексе для R изомера и запустить минимизацию повторно. А для S изомера с новыми параметрами сделать комплекс и далее по списку.
#39
Updated by Anastasia Rachkova almost 4 years ago
Переделала все еще раз, подкорректировала воду, в который сомневалась в прошлый раз. Но снова при запуске gmx mdrun -v -deffnm em все ломается.
step 11: One or more water molecules can not be settled.
Check for bad contacts and/or reduce the timestep if appropriate.
Back Off! I just backed up step11b_n15.pdb to ./#step11b_n15.pdb.1#
Back Off! I just backed up step11c_n14.pdb to ./#step11c_n14.pdb.1#
Back Off! I just backed up step11c_n15.pdb to ./#step11c_n15.pdb.1#
Wrote pdb files with previous and current coordinates
tMPI error: Receive buffer size too small for transmission (in valid comm)
Aborted (core dumped)
Файл с новыми координатами стопорится на одной из добавленных молекул воды, ничем не примечательной:
ATOM 23147 OW SOL 1 50.713 47.831 12.344 1.00 0.00
ATO
Может, стоит еще уменьшить размер ячейки (чтобы минимизировать систему меньшего размера)? Или стоит просмотреть все файлы с новыми координатами в паймоле (интересно, что у меня не получилось пока их открыть) и искать глазами, где что не так встало?
#40
Updated by Anastasia Rachkova almost 4 years ago
Для S-изомера все получилось, с ним работала в новой папке
/home/domain/rachkovanastya/PGM_S_start